X-HTVS - "Screen millions of compounds in one go"
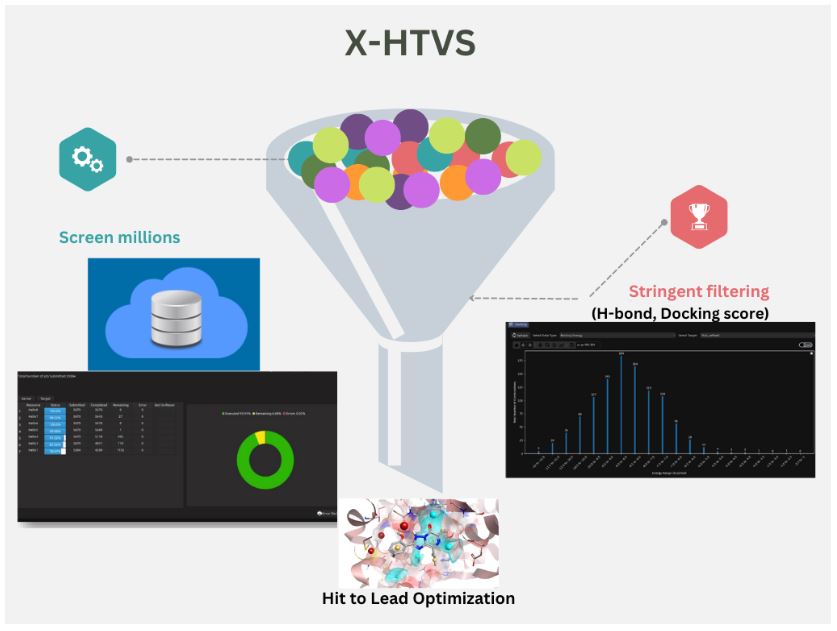
In recent times, Molecular Docking has become a crucial step in drug discovery and design, providing a deeper insight into the orientation and stability of a target molecule with a ligand. X-HTVS supports multi-target multi-ligand screening, i.e., screening of any number of ligands against multiple targets simultaneously. Our application provides the user with one of the best possible docking methods currently available for hit to lead identification. Combining this with further refinements in sampling and thoroughness, it makes the whole virtual drug screening process much faster and computationally less expensive. The app's ability to link the screening platform to one or more high performance computing (HPC) resources, independent workstations on the same network, and cloud servers via the integrated data connector (DC) of PRinS3 is another one of its distinguishing features. This makes it possible to divide the jobs among multiple threads, giving them a considerable speed boost. It is by taking advantage of the same attributes we can assume, technically, there should be no limits to the number of combinations attempted, giving the app a significant edge over competing platforms.
Exploiting the power of this application, our in-house team has successfully produced findings for data sets of up to 7.5 lakh protein-ligand combinations (0.7 million) with great accuracy, within a record amount of time. The outcomes from the dataset of a million compounds acquired from the Molport portal are also in the works at the time of the publication of this article. Prescience Insilico strives to bring the transformations in the drug discovery scenario to the fingertips of everyone interested. The super straightforward interface of X-HTVS lets the users achieve their desired results even with the most fundamental comprehension of the process. It is possible to switch between preferred algorithms (Lamarckian genetic algorithm or local search), model types (bound, extended), or even the kind of GA runs (coarse grain, highly coarse grain, fine-grain and user-defined), giving complete control to the users. Multiple input file formats are accepted, adding to the versatility. There is also a time estimation feature, where users can get an estimate about how long it could take to perform their chosen docking jobs, even before starting the runs. Simple histogram representations of binding energy, binding score and number of H-bonds are offered for quick analysis of the results. Users can download them in either a summarized or detailed format, as they please. The downloaded docked complex structures can further be subjected to MD simulations using them as inputs in the X-ESS application of PRinS3 platform.
Explore X-HTVS and expose yourselves to the innovations of virtual drug screening with its untapped potential. For access to the platform and all the information needed for the optimal experience, please reach out to us at support@prescience.in. For a holistic framework of the complete drug discovery pipeline, visit www.prescience.in and browse through our existing available applications, including BioIn, ChemIn, X-ESS and SyMoG.
Lobelia Ghosh