The traditional procedure of discovering new drugs is challenging and time-consuming, often having very low success rates. To date, scientists have to go through a very tedious process of research, selection and synthesis of drug candidates before they can be tested in multiple levels of pre-clinical and clinical trials. However, with the rising wave of new technology, in silico (computational) modeling is starting to take center stage in drug design and discovery.. Prescience Insilico(PRinS3) is a forerunner in utilizing the power of computation (on premise HPC and Public Cloud) and artificial intelligence to turn the novel drug screening pipeline into an easy, efficient, economical and rapid ordeal.
Prescience Insilico has brought forward a modern integrative computational platform PRinS3 which comprises five separate Applications (App), namely BioIn, ChemIn, X-ESS, AI/SyMoG and X-HTVS. These APPs provide the core components required for smooth and systematic virtual drug screening. X-ESS comprises modules like molecular docking, molecular dynamics simulations (MD), MM-PBSA and Metadynamics for estimating the free energy barriers of binding ligands to targets in solvents like water. With a constant network of effort, the company has been working on a series of other Apps as well to provide the researchers with an all-inclusive experience of exploration. At the moment, X-ESS can manage large datasets (of around 500 combinations; i.e. multi target and multi ligand) for screening at a time and operates various stages of the process by connecting the software to the best hardware resources available. However, all of these require a considerable amount of processing power, in some cases a local workstation may fall short. Thus, other than the local machine, the software also has the Data-connector helpful in connecting to any high performance computing (HPC) environment (e.g. National Super Computing Mission - NSM at Indian Institute of Technology Kanpur) or public cloud platforms (e.g. Google Cloud), based on the user’s requirements and choice. In this study, a system made up of protein-ligand complexes was screened on different hardware platforms i.e., workstation, HPC at NSM - IITK, Google Cloud Platform) in order to evaluate the performance and efficiency of the X-ESS across variable computing hardware. One could run X-ESS and X-HTVS apps on a hybrid platform as well.
Choice of DatasetPAK1 or p21-activated kinase 1 is a pharmacologically important protein because of its intense involvement in a multitude of signaling pathways responsible for various cellular processes. Mutations/overexpression of this gene can lead to diseases like IDDMSSD (Intellectual developmental disorder with macrocephaly, seizures, and speech delay) and also a variety of cancers. For this study, along with the wild type protein, four mutant variations were also used which have mutations at 299, 389, 393 and 423th residues.
Significance of the selected mutations are outlined in the following table.
| Mutated Residue Number | Significance of the Mutation |
|---|---|
| 299 (Lys->Arg) | Decrease and abolition of kinase function |
| 389 (Asp->Asn) | Can cause abolition of kinase activity |
| 393 (Asp->Ala) | Dissolves the ability to auto-phosphorylate at Thr-423 |
| 423 (Thr->Glu) | Constitutive kinase activity and decrease of CDC42-stimulated activity |
Computational Resources
GROMACS 2018.3 was built in single precision with GCC 8.3.0, FFTW 3.3.8 (single precision), OpenMP multithreading and GPU was supported with CUDA 10.2. Plumed 2.5.4 was patched with gromacs. Autodock 4.2.6 with MGLTools 1.5.7 was used for Docking studies. We used docker containers to deploy these packages on the host machines (GC and Workstations). The docker containers had everything the PRinS3 applications needed to run including libraries, system tools, code, and runtime. The advantage of using Docker is that we can quickly deploy and scale applications into any environment and gives us the confidence that our code will run. This creates a highly reliable, low-cost way to build, ship, and run distributed applications at any scale. We have built our docker container on Debian OS harnessing the GPU power. It can be deployed on any Linux, Windows, or macOS computer.
Technical Specifications of Machine Used in this Study
| Machine | Processor | Sockets X cores | Threads/cores | Clock (GHz) | GPUsnVidia |
|---|---|---|---|---|---|
| NSM | Intel Xeon | 2 x 20 | 1 | 2.5 | V100 |
| Workstation | i9-9820X | 1 x 10 | 2 | 3.30 | GeForce RTX 2080 Ti |
| GC | Intel Xeon | 1 x 4 | 2 | 2.0 | Tesla T4 |
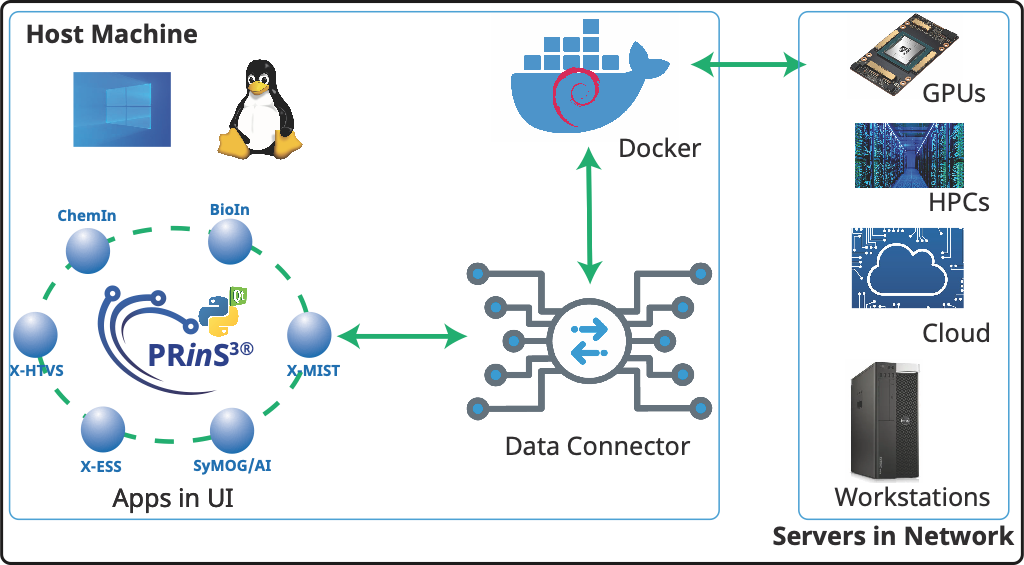
Figure 1. PRinS3 setup and its communication with the servers in the network. The UI is installed on the host’s desktop (or laptop). The UI bundles the applications and data connector (mechanism to connect to the servers). Along with the UI, a docker image is deployed as a container in the host desktop (or laptop). This host machine can also be used for performing the computations.
Testing the power of computational resources
The Benchmark dataset contains 255 protein-ligand combinations made up of high resolution crystal structure of refined and processed PAK1 kinase protein (PDB ID: 3Q52), its four mutants, along with 51 kinase approved drugs for repurposing. The refinement of the target proteins and retrieval of the data for FDA approved drugs were done using the PRinS3 software itself.
Each step of the screening is primarily divided into three sections, i.e., Preprocessing and upload of data, Job run and Downloading the results. At first, the docking of each ligand into the corresponding binding pockets of the targets was ensured by providing the binding residue numbers ; i.e., 299 and 389. The number of Genetic Algorithm (GA) runs was set to 100; default for our system. After the completion of preprocessing, the files were uploaded to the selected server by data connector (proprietary tool developed by Prescience Insilico) and docking jobs were run. The calculations of molecular docking in X-ESS are done using Autodock 4 and the docking is carried out based on Lamarckian genetic algorithm. Upon download and analysis of the data derived from molecular docking, 25 protein-ligand combinations were funneled out to take forward for MD. Advanced parameters were kept at their defaults for the operations and the NPT/NVT simulation time was set at 5ns. To simulate MD, water molecules were added to the system in the form of a water model. The estimation of size of the whole system in terms of number of atoms is provided below.
System Details
| Component | No of Atoms |
|---|---|
| Protein | -4640+/-3 |
| Ligand | -70+/-4 |
| Water | -91005+/-10 |
The time taken for preprocessing, simulation and data download of each step was noted and used as a gauge of performance of the PRinS3 software. The procedure was repeated in the exact same manner across three different resources; i.e., NSM HPC, cloud and local workstation. The data derived from each case were analyzed and compared with each other to reflect on the contribution of computational resources in acceleration of the processes.
Result and Discussion
In order to provide a simplistic outlook on the efficiency of PRinS3, we looked into and recorded the duration of preprocess/upload, runtime and data download for Molecular docking, MD and FES each; across each of the three different servers.
When working on a local workstation, the preprocessing and upload of our system files to the server in case of docking, MD and FES took nearly 33+/-1 seconds, 22+/-1 seconds and 23+/-0.5 seconds respectively. When using a NSM server, the docking, MD and FES took around 36+/-2, 26+/-5 and 25.5+/-0.5 seconds respectively. In case of a cloud, the upload time for docking, MD and FES were found to be 41+/- 0.5, 37+/-1 and 37+/-4 seconds respectively
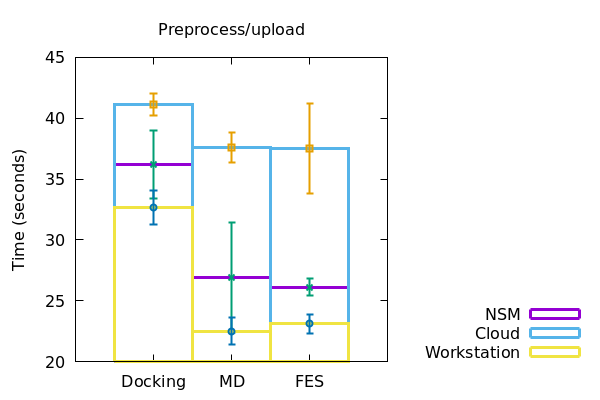
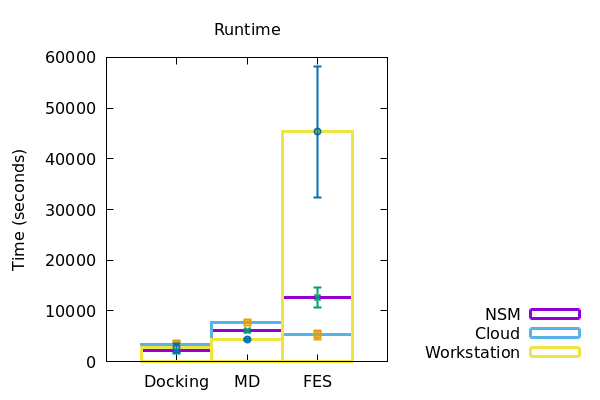
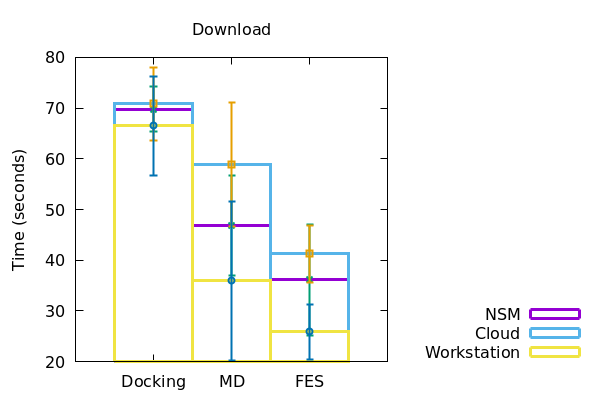
Figure 2
In a similar fashion, the duration of runtime of jobs and download of files to the local machine were also recorded and plotted accordingly (Figure 2). The data derived has been represented in a tabular form below.
| Computational resources | Stage of screening | Preprocessing/Upload (s) | Runtime (s) | Download (s) |
|---|---|---|---|---|
| Docking | 33+/-1 | 3000+/-500 | 57+/-10 | |
| Workstation | MD | 22+/-1 | 5000+/-50 | 36+/-16 |
| FES | 23+/-0.5 | 45000+/-13000 | 26+/-6 | |
| Docking | 36+/-2 | 2000+/-500 | 70+/-5 | |
| NSM Server | MD | 26+/-5 | 7000+/-50 | 47+/-10 |
| FES | 25.5+/-0.5 | 12000+/-2000 | 37+/-11 | |
| Docking | 41+/- 0.5 | 4000+/-500 | 71+/-1 | |
| Cloud | MD | 37+/-1 | 8500+/-100 | 59+/-12 |
| FES | 37+/-4 | 6000+/-1000 | 42+/-5 |
Conclusion
Docking with the same dataset, the time taken for preprocessing and data download took increasingly more time for workstation, NSM server and google cloud respectively. However, for running the docking jobs itself, the NSM server took the least amount of time, the cloud taking the most. In the case of MD however, the workstation performed all of the preprocessing, run and download in the least amount of time. The NSM server being the next, and the cloud following soon after. Interestingly, at the time of FES runs, the workstation consumed considerably more time than both NSM server and the cloud server, making NSM the most efficient for the job at hand. Although for preprocessing and downloading of data from FES, the time increased from workstation to NSM server and then to cloud.
Lobelia Ghosh