Refereed Journals
Note: PDFs are provided for personal use only, for non-commercial research and education. It should not be retransmitted,redistributed or uploaded to any public server, network, bulletin board, online service without permissions from the publisher and author. You may not make copies for any commercial purpose. Reproduction or storage of materials retrieved from this website is subject to the Copyright Act.
2023 |
|
Abstract: Pancreatic lipase (PL) is a keen target for anti-obesity therapy that reduces dietary fat absorption. Here, we investigated the binding patterns of 220 PL inhibitors having experimental IC50 values, using molecular docking and binding energy calculations. Screening of these compounds illustrated most of them bound at the catalytic site (S1-S2 channel) and a few compounds are at the non-catalytic site (S2-S3 channel/S1-S3 channel) of PL. This binding pattern could be due to structural uniqueness or bias in conformational search. A strong correlation of pIC50 values with SP/XP docking scores, binding energies (ΔGMMGBSA) assured the binding poses are more true positives. Further, understanding of each class and subclasses of polyphenols indicated tannins preferred non-catalytic site wherein binding energies are underestimated due to huge desolvation energy. In contrast, most of the flavonoids and furan-flavonoids have good binding energies due to strong interactions with catalytic residues. While scoring functions limited the understanding of sub-classes of flavonoids. Hence, focused on 55 potent PL inhibitors of IC50 < 5 µM for better in vivo efficacy. The prediction of bioactivity, drug-likeness properties, led to 14 bioactive compounds. The low root mean square deviation (0.1-0.2 nm) of these potent flavonoids and non-flavonoid/non-polyphenols PL-inhibitor complexes during 100 ns molecular dynamics runs (MD) as well as binding energies obtained from both MD and well-tempered metadynamics, support strong binding to catalytic site. Based on the bioactivity, ADMET properties, and binding affinity data of MD and wt-metaD of potent PL-inhibitors suggests Epiafzelechin 3-O-gallate, Sanggenon C, and Sanggenofuran A shall be promising inhibitors at in vivo conditions.
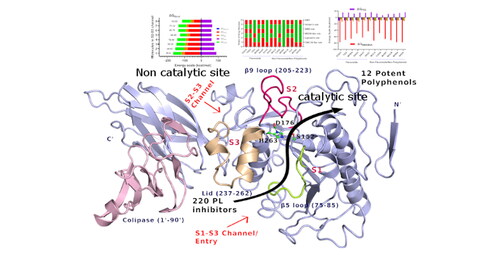
|
|
|
2022 |
|
Abstract: We investigate the concentration-dependent phase diagram of pluronic L64 in aqueous media at 300 and 320 K using coarse-grained (CG) molecular dynamics (MD) simulations. The CG model is derived by adapting the Martini model for nonbonded interactions along with the Boltzmann inversion (BI) of bonded interactions from all-atom (AA) simulations. Our derived CG model successfully captures the experimentally observed micellar-, hexagonal-, lamellar-, and polymer-rich solution phase. The end-to-end distance reveals the conformational change from an open-chain structure in the micellar phase to a folded-chain structure in the lamellar phase, increasing the orientational order. An increase in temperature leads to expulsion of water molecules from the L64 moiety, suggesting an increase in L64 hydrophobicity. Thermodynamic analysis using the two-phase thermodynamics (2PT) method suggests the entropy of the system decreases with increasing L64 concentration and the decrease in free energy (F) with temperature is mainly driven by the entropic factor (−TS). Further, the increase in aggregation number at lower concentrations and self-assembly at very high concentrations is energetically driven, whereas the change from the micellar phase to the lamellar phase with increasing L64 concentration is entropically driven. Our model provides molecular insights into L64 phases which can be further explored to design functionality-based suprastructures for drug delivery and tissue engineering applications.
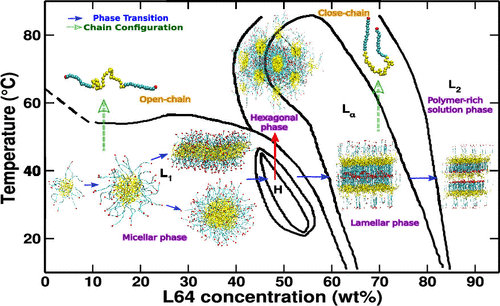
|
|
Abstract: In recent times, computational methods played an important role in the down selection of chemical compounds, which could be a potential drug candidate with a high affinity to target proteins. However, the screening methodologies, including docking, often fails to identify the most effective compound, which could be a ligand for the target protein. To solve that, here we have integrated meta-dynamics, an enhanced sampling molecular simulation method, with all-atom molecular dynamics to determine a specific compound that could target the main protease of novel severe acute respiratory syndrome coronavirus 2 (SARS-COV-2). This combined computational approach uses the enhanced sampling to explore the free energy surface associated with the protein's binding site (including the ligand) in an explicit solvent. We have implemented this method to find new chemical entities that exhibit high specificity of binding to the 3-chymotrypsin-like cysteine protease (3CLpro) present in the SARS-CoV-2 and segregated to the most strongly bound ligands based on free energy and scoring functions (defined and implemented) from a set of 17 ligands which were prescreened for synthesizability and druggability. Additionally, we have compared these 17 ligands' affinities against controls, N3 and 13b α-ketoamide inhibitors, for which experimental crystal structures are available. Based on our results and analysis from the combined molecular simulation approach, we could identify the best compound which could be further taken as a potential candidate for experimental validation.
|
|
|
2021 |
|
Abstract: Since the onset of global pandemic, the most focused research currently in progress is the development of potential drug candidates and clinical trials of existing FDA approved drugs for other relevant diseases, in order to repurpose them for the COVID-19.
|
|
|
2020 |
|
Abstract: A hybrid approach combining machine learning algorithms with molecular simulation is utilized to screen hypothetical metal–organic framework (h-MOF) database for the best material to separate ethane (C2H6) and ethylene (C2H4).
|
|
Abstract: Here, we report new chemical entities that exhibit highly specific binding to the 3-chymotrypsin-like cysteine protease (3CLpro) present in the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
|
|
|