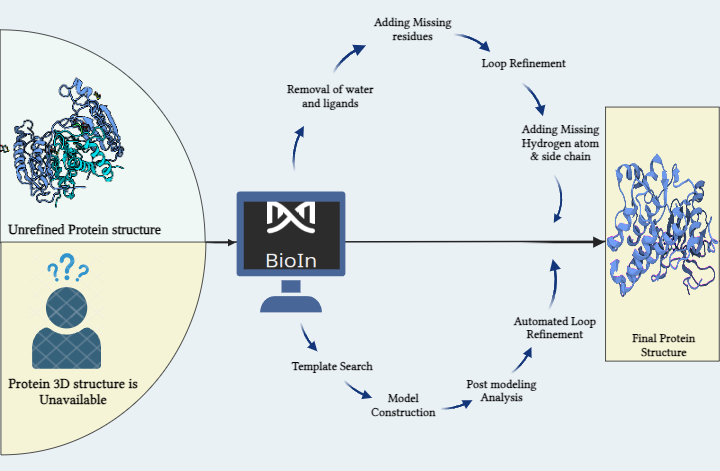
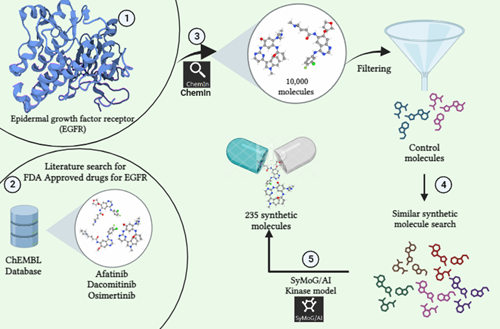
Drug discovery is a process of uncovering new potential candidates for disease treatment. Unfortunately, the overall success rate of drug development is only 1 - 4 % with 90% failure in clinical developmental stages. Their inadequate ADMET qualities being the root cause of the majority of failures. Additionally, the process is demanding, spanning a long 9-12 years. Today, novel drug discovery research takes approximately ~1 billion dollars which largely includes the cost of chemical synthesis of compounds and preclinical as well as clinical trials. Even though there are massive capital investments in pharmaceutical companies, the significant innovation gap is still prominent. From 1985-2020, FDA has approved only 1149 new molecular entities (NME) or new drugs which endorses the notion that traditional drug discovery is indeed an inefficient process. With exponential growth of technology, Artificial intelligence has revolutionized the drug discovery process by minimizing the risk of failure. AI is a computer technology-based system with algorithms or tools which mimic human intellect. They are trained to learn from previously-known data, make decisions and predict the outcome for the unknown. AI models efficiently handle gigantic amounts of data & massive calculations, enhancing the automation. It can be beneficial in designing clinical trials (patient selection, patient drop-out) and monitoring, automated manufacturing of drugs, market research and predictions, bioactivity/toxicity/physicochemical property predictions etc. AI can quickly search the vast chemical space for targets and provide quantitative recognition of hit and lead compounds. Since AI is trained on the input data, its success solely depends on them. Nevertheless, AI appears to be a boon to drug research.
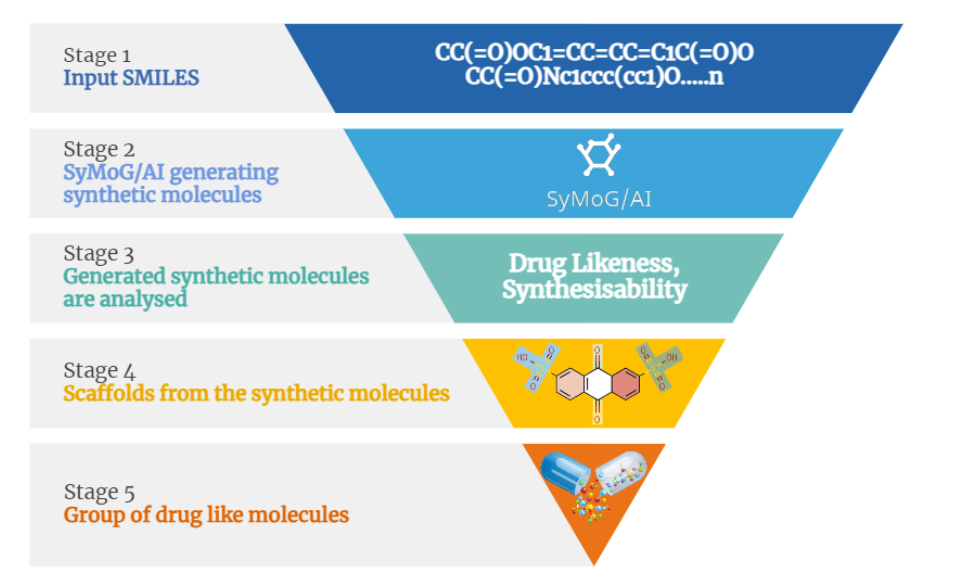
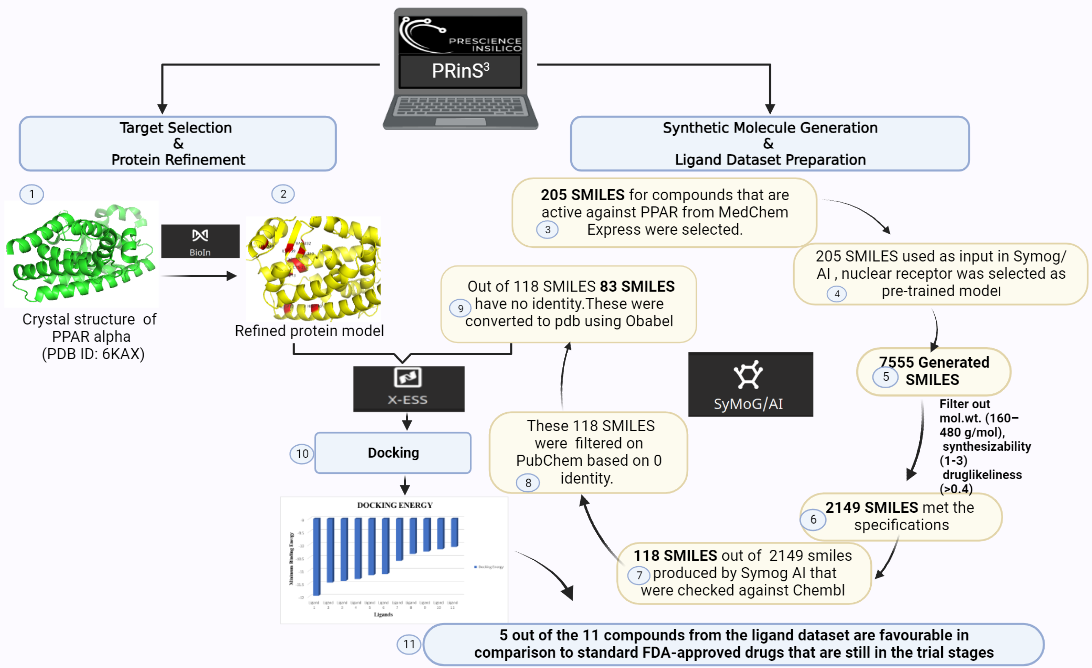
Here at Prescience Insilico, we are developing algorithms to design solutions to this problem. We came up with an App, AI/SyMoG (
Artificial
Intelligence/
Synthetic
Molecule
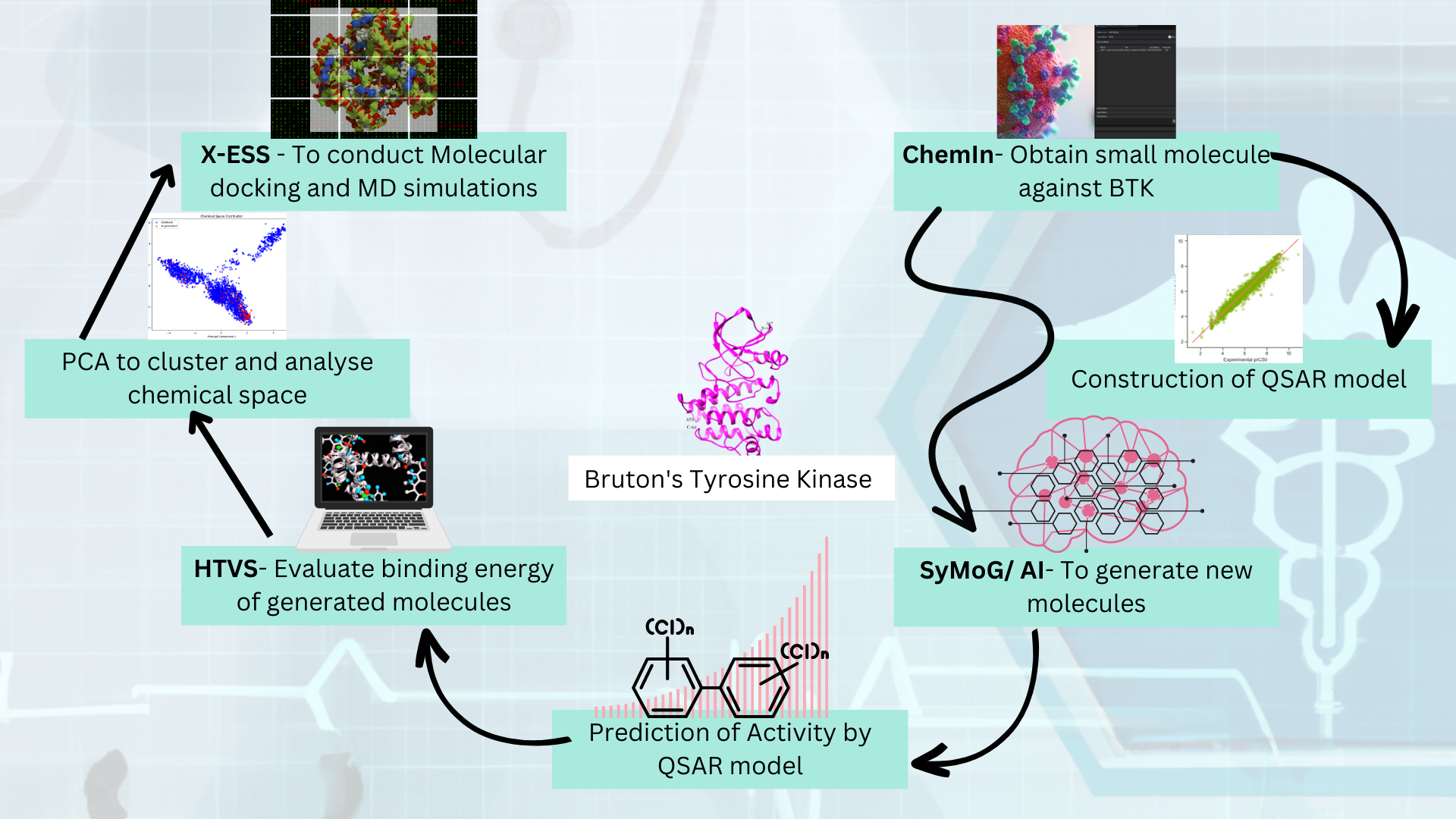
Generator) as a module in the PRinS3 platform that is trained on scaffolds from input using a graphical neural network. Key features of our app is we have models trained based on different protein classes ie kinases, phosphates, transferases to name a few. Further the App provides more information on physicochemical properties of the input molecules and new molecules generated. We can also differentiate between the molecules generated from the input by looking at their scaffold from the scaffold clustering being computed. These molecules can be visualized and checked for their similarities. The large data could also be filtered and downloaded as PDB, which can be directly used for X-HTVS or X-ESS as potential ligands for screening. We carefully and tactically designed this app, to help in finding novel molecules that can be potent drug candidates. This also helps in identifying the novel molecules and input scaffold based drugs. So, scientists can file an IP and create a large database of molecules, which could be further validated by our virtual screening App X-HTVS and X-ESS. For more details of these Apps please visit our webpage at
https://www.prescience.in/prins. For accessing the platform and all the information needed for the optimal experience, please reach out to us at
support@prescience.in
For a holistic framework of the complete drug discovery pipeline, visit
www.prescience.in and browse through our existing available applications, including BioIn, ChemIn, X-ESS and X-HTVS.
Sumedha Bhosale