Streamlining MM-PBSA Workflows with X-ESS: Automation Meets High-Performance Computing
Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) is a widely used computational method in drug discovery to estimate binding free energies between receptors and ligands. It combines molecular mechanics energy (ΔEMM), polar solvation energy (ΔEPB), non-polar solvation energy (ΔESA), and entropic contributions (TΔS) to calculate the binding free energy (ΔGbind) of a ligand-protein complex. The binding free energy is calculated as:
ΔGbind = ΔEMM + ΔEPB + ΔESA − TΔS
Binding free energy (ΔGbind) measures the stability of a ligand-protein complex relative to its unbound components, providing valuable insights into the strength and nature of molecular interactions. This information is crucial for optimizing drug candidates during the development process. However, the accuracy of MM-PBSA results depends significantly on the choice of force fields, which can yield varying results due to differences in parameterization. X-ESS overcomes this challenge by offering multiple force-field options, including SwissParam (CHARMM27), LigParGen (OPLS-AA), and user-defined parameters. This flexibility enables researchers to tailor their simulations to balance computational efficiency and precision.
Using MD Simulations and MM-PBSA to Optimize Kinase Inhibitors for Cancer Treatment
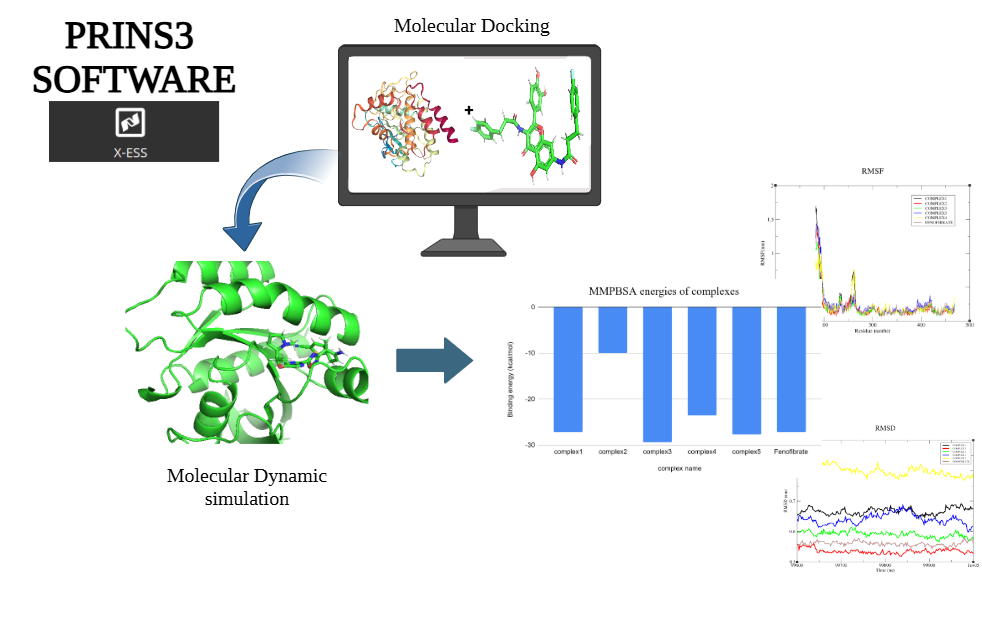
When developing a kinase inhibitor as a potential cancer treatment, computational methods play a critical role in understanding and optimizing molecular interactions. Here's an example of how molecular dynamics (MD) simulations combined with MM-PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) analysis can guide drug design. From Docking to Dynamics: The process begins with docking, which predicts the initial binding complex between the kinase protein and its inhibitor. However, proteins are not rigid structures, and the ligand's mobility within the binding site needs to be accounted for. To address this, the docked complex undergoes a 50-nanosecond MD simulation, allowing the system to explore its conformational flexibility. Capturing Key Interactions: Throughout the simulation, 200 snapshots of the complex are taken at regular intervals to capture different conformational states. MM-PBSA analysis is then applied to these snapshots to calculate the binding free energy (ΔGbind). In this case, the binding energy was ~-40 kcal/mol, indicating a strong affinity between the kinase and the inhibitor. This is driven by favorable van der Waals and electrostatic interactions, balanced by minimal desolvation penalties and optimized hydrophobic contributions. Reducing entropic penalties through rigid, well-fitting ligands further enhances binding affinity. We can improve binding affinity and specificity by exploring key interactions within the binding region.
Traditional MM-PBSA procedures often involve multiple tools and manual steps, creating opportunities for errors and inconsistencies. These challenges can slow down the workflow and impact the reliability of results, especially in complex or large-scale studies. X-ESS, a revolutionary platform designed to fully automate the MM-PBSA workflow. From molecular docking and MD simulations to energy computations, X-ESS eliminates the need for manual intervention at every stage. Built for High-Performance Research: X-ESS integrates with high-performance computing (HPC) resources, enabling rapid data analysis and computational tasks. This capability drastically reduces the time required for energy calculations, making it an ideal tool for large-scale research projects and drug discovery pipelines. Transforming Computational Chemistry: With its automated and scalable approach, X-ESS is transforming how researchers conduct MM-PBSA analyses, paving the way for faster, more accurate insights into molecular interactions. Whether you're optimizing a lead compound or conducting a broad compound screening, X-ESS simplifies the process while delivering robust and reliable results.
For more information on the X-ESS module and the PrinS3 software suite, visit our official website at https://prescience.in/. To access the platform or receive personalized assistance, contact us via email at support@prescience.in. Learn more about PrinS3 and its applications at PrinS3 Software. With the integration of MM-PBSA in X-ESS, PrinS3 is setting new standards in computational drug discovery, providing researchers with tools to overcome traditional challenges and drive innovation in the discovery of new therapeutics.Additionally, you can find valuable information on our applications at https://prescience.in/prins3/